Заявка на патент РФ № 2000122958/04(024306)
ЗАЯВКА НА ПАТЕНТ РФ №2000122958/04(024306) СПОСОБ ОКСИСЛЕНИЯ ПРОСТРАНСТВЕННО ЗАТРУДНЕННЫХ ФЕНОЛОВ
Заявитель: А.Г.Ахмадуллина
Авторы: А.Г.Ахмадуллина, Н.А.Мукменева, В.Х.Кадырова, С.В.Бухаров, Л.К.Фазлиева, Г.М.Нургалиева, А.С.Шабаева
Изобретение относится к способам получения замещенных дифенохинонов и бисфенолов, которые могут быть использованы в химической, нефтехимической, газовой, нефтеперерабатывающей и пищевой отраслях промышленности в качестве реагентов органического синтеза, стабилизаторов полимерных материалов (полиэтилена, полипропилена, натурального и синтетического каучуков), антиокислительных присадок (АОП) к смазочным маслам, моторным топливам, олефинам, животным и растительным жирам.
Известен способ окисления пространственно-затрудненных фенолов, прежде всего 2,6-дитрет-бутил-фенола, кислородом воздуха в присутствии комплексного гомогенного катализатора: солей меди с аминами /1/. Недостатками этого способа являются: загрязнение получаемого дифенохинона и конечного продукта окисления — 4,41-бис(2,6-дитрет-бутил-фенола) примесями солей меди, сильно снижающими его стабилизирующую эффективность; непрерывное расходование гомогенного катализатора окисления и образование большого обьема токсичных стоков из-за трудности выделения гомогенного катализатора из отработанных растворителей и промывных вод. Поэтому указанный способ до сих пор не внедрен в промышленность, несмотря на острую потребность в упомянутом антиоксиданте.
Известны также методы окисления замещенных фенолов кислородом воздуха в присутствии фталоцианинов и салькоминов металлов /2,3/. Недостатками этих способов являются низкая селективность процесса, сопровождающегося образованием смесей дифенохинона (26%) и бензохинона (73 %); большой расход гомогенного катализатора, растворенного в полярных растворителях — амидах, сульфоксидах или спиртах С1-С4.
Известен также метод окислительного сдваивания пространственно затрудненных монофенолов, например 2,6-дитретбутил-фенола, в соответствующий дифенохинон (ДФХ) при контакте с кислородом воздуха в присутствии щелочного катализатора – гидроксида щелочного металла /4/. Затем в реактор с ДФХ в атмосфере инертного газа добавляют свежую порцию монофенола, который при нагревании в присутствии щелочного катализатора взаимодействует с ДФХ с образованием соответствующего бисфенола. Недостатками этого способа являются недостаточно высокие выхода ДФХ и целевого БФ, необходимость обработки реакционной массы (после завершения реакции) кислотами и водной отмывки синтезированных продуктов от кислот и щелочей с образованием сточных вод.
По технической сущности и достигаемому результату наиболее близким к предлагаемому является процесс синтеза 4,41-бис(дитрет-бутил-фенола) через промежуточную стадию синтеза ДФХ окислением вышеупомянутого пространственно затрудненного бисфенола (БФ) воздухом при 30-300оС и давлении 3.5-35 атм. в среде растворителей с Ткип. от 80 до 200оС (лучше в трет.-бутиловом спирте, толуоле или диэтилбензоле) в присутствии катализаторов, которыми могут служить гидроксиды и соли щелочных и щелочно-земельных металлов или их водные растворы, аминные основания, хелаты металлов переменной валентности либо смесь указанных видов катализаторов. Предпочтительным является использование дешевых и доступных гидроксидов щелочных металлов /5/.
После полного окисления БФ с образованием ДФХ на 1 стадии процесса прекращают подачу воздуха, реактор охлаждают, стравливают из него давление и продувают систему инертным газом. На 2 стадии синтеза БФ, заключающемся в дегидрировании 2,6-дитретбутилфенола (2,6-ДТБФ) полученным ДФХ, в реакционную смесь с ДФХ добавляют соответствующий монофенол и полученную смесь нагревают до 100-350оС, выдерживая при за-данной температуре 1-4 часа (в зависимости от температуры). Затем щелочной катализатор нейтрализуют, добавляя в реактор кислоту, чаще ортофосфорную. После охлаждения реакционной смеси отфильтровывают выпавшие желтые кристаллы БФ и подвергают их водной отмывке от кислоты и перекристаллизации из растворителя, например из толуола, эфира или спирта. Часть полученного БФ рециклизуют на стадию синтеза ДФХ. Недостатками прототипа являются: использование повышенных давлений и недостаточно высокий выход ДФХ на стадии окисления пространственно затрудненного БФ, а также необходимость постоянного и полного обновления щелочного катализатора, нейтрализуемого кислотами на 2 стадии синтеза при выделении целевого БФ, и образование сточных вод.
Задачей настоящего изобретения является устранение указанных недостатков за счет совершенствования технологии стадии окисления пространственно затрудненных фенолов с получением ДФХ. Согласно предлагаемому изобретению окисление пространственно затрудненных фенолов кислородом воздуха проводят при нагревании до 70-90оС и давлении 0.1-1.0 МПа в растворителях в присутствии окислительно-каталитической системы, состоящей из хелата металла переменной валентности на полимерном носителе и/или промотора и инициатора окисления.
Реакционный раствор ДФХ, полученный после окисления пространственно затрудненных моно- или бисфенолов в вышеописанных условиях, в горячем состоянии отделяют от стационарного слоя катализатора и отстаивают. Отстоявшийся верхний углеводородной слой с ДФХ отделяют от промотора, охлаждают и отфильтровывают выпавшие кристаллы ДФХ, а промотор и фильтрат растворителя с остаточным количеством ДФХ рециркулируют в реактор с катализатором на стадию синтеза ДФХ. Стадию получения целевого БФ дегидрированием 2,6-дитретбутил-фенола синтезированным ДФХ осуществляют одним из известных способов, например, по способу /6/ растворением смеси ДФХ с 2,6-ДТБФ в диметилформамиде и выдержкой реакционной смеси при 120-150оС в течение 5-6 часов, отфильтровыванием кристаллов БФ от охлажденного растворителя с последующей рециркуляцией фильтрата на стадию синтеза БФ.
В качестве хелата металла в предлагаемом способе используют фталоцианин кобальта и/или другие водонерастворимые каталитически активные компоненты, жестко закрепленные на полимерном носителе катализатора. В качестве промотора окисления используют продукт превращения кислых примесей высококипящих углеводородных фракций образующийся в процессе их гетерогенно-каталитической очистки от меркаптанов и кислых примесей обработкой кислородом воздуха в щелочной среде. Промотор окисления (Пж) представляет собой вязкую жидкость красновато-коричневого цвета с плотностью не менее 1,0 кг/л, нерастворимую в углеводородных растворителях и обладающую деэмульгирующими свойствами, вследствие чего легко и полно отделяющуюся от них простой декантацией. В качестве инициатора окисления используют дифенохинон (ДФХ), получаемый при окислении пространственно затрудненных моно- и бисфенолов, который либо специально подают в реактор окисления (на первоначальном этапе синтеза), либо он поступает в составе рециркулирующего растворителя, отфильтрованного от кристаллов синтезированного ДФХ.
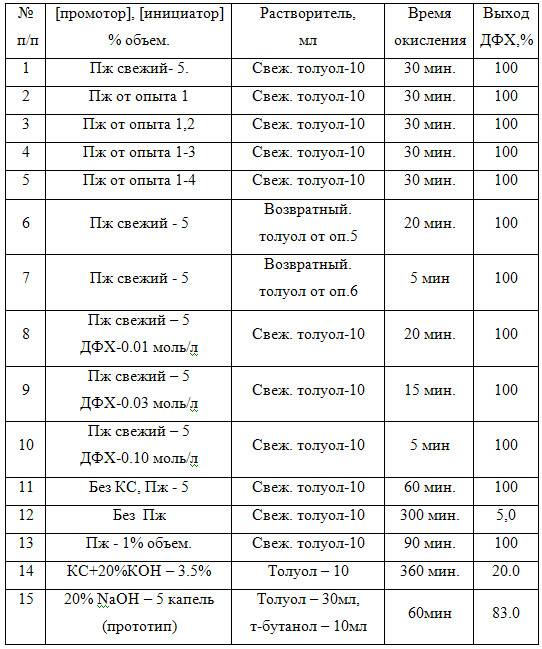
Таким образом, отличительным признаком предлагаемого способа является использование при окислении пространственно затрудненных фенолов вышеуказанной окислительно-каталитической системы, состоящей из гетерогенного катализатора на полимерной основе и/или промотора и инициатора окисления. Данный отличительный признак предложенного способа определяет его существенные отличия в сравнении с прототипом и известным уровнем техники в данной области, т.к. использование указанной каталитической системы для окисления пространственно затрудненных фенолов в литературе не описано и позволяет, по сравнению с прототипом, снизить температуру и давление, повысить скорость окисления замещенных моно- и бисфенолов, увеличить выход ДФХ, значительно снизить потери и повысить срок службы компонентов каталитической системы. Предлагаемый способ апробирован в лабораторных условиях при окислении 4,41-бис(2,6-дитрет-бутил-фенола) — БФ (примеры 1-15 и табл.1), и 2,6-дитрет-бутил-фенола — 2,6-ДТБФ (примеры 16-22 и табл.2) кислородом воздуха в различных растворителях в присутствии фталоцианиновых катали-заторов КС/7/ и КСМ /8/ на полипропилене. В качестве промотора (Пж) в опытах был использован продукт превращения кислых примесей керосиновой фракции, образовавшийся в процессе ее окислительно-каталитической демеркаптанизации обработкой кислородом воздуха в щелочной среде /9/.
Пример 1.
В емкость для окисления (реактор) загружают 1,3 г катализатора КС в виде пластинок размером 2х2 мм, 10 мл толуола, 0,5 мл (5% объем.) Пж и 1г (0.0025 моля) БФ. При температуре 85оС через реакционную смесь барботируют воздух до полного исчерпывания исходного БФ. Контроль за ходом процесса осуществляют с помощью тонкослойной хроматографии на силуфоловых пластинках в системе гексан:ацетон=9:1. Верхний углеводородный слой отстоявшегося горячего реакционного раствора с ДФХ отделяют от каталитической системы (от катализатора КС и промотора Пж), осторожно сливая его в стакан, в котором, по мере охлаждения, выпадают фиолетовые кристаллы ДФХ. Последний отфильтровывают от растворителя (толуола) и получают 1гДФХ с Тпл=246оС, что соответствует литературным данным и составляет 100% от теоретического выхода.
Пример 2.
По примеру 1 окисляют новую порцию (0,0025 моля) БФ в 10 мл свежего толуола в течение 30 минут в присутствии каталитической системы, использованной в примере 1 (оставшейся в реакторе после слива углеводородной фазы). Выделен 1г (100%) ДФХ.
Пример 3.
По примеру 1 окисляют 0,0025 моля БФ в 10 мл свежего то-луола в присутствии каталитической системы, использованной в примерах 1 и 2. Через 30 мин. БФ исчерпался. Выделен 1г (100%) ДФХ.
Пример 4.
По примеру 1 окисляют 0,0025 моля БФ в 10 мл свежего то-луола в присутствии каталитической системы, использованной в примерах 1-3. Через 30 мин. окисления выделен 1г (100%) ДФХ.
Пример 5.
По примеру 1 окисляют новую порцию (0,0025) моля БФ в 10 мл свежего толуола (0.25 моль/л) в присутствии каталитической системы, использованной в примерах 1-4. Выделено 1г (100%) ДФХ.
Пример 6.
По примеру 1 окисляют 0,0025 моля БФ в 10 мл возвратного толуола, отфильтрованного от кристаллов ДФХ в примере 5. Полное исчерпывание исходного бисфенола БФ происходит через 20 минут.
Пример 7.
По примеру 1 окисляют 0,0025 моля БФ в 10 мл возвратного толуола от примера 6. Полное исчерпывание исходного бисфенола БФ с образованием 100% ДФХ происходит через 5 минут.
Пример 8.
По примеру 1 окисляют 0,0025 моля БФ в 10 мл свежего толуола с добавкой в реакционную смесь 0,0442г (0.01 моль/л) ДФХ. Полное исчерпывание исходного бисфенола БФ с образованием ДФХ происходит через 20 минут окисления кислородом воздуха.
Пример 9.
По примеру 1 окисляют 1г (0,25 моль/л) БФ в 10 мл свежего толуола с добавкой в реакционную смесь 0,1363 г (0.031 моль/л) ДФХ. Полное исчерпывание исходного БФ происходит через 15 минут.
Пример 10.
По примеру 1 окисляют 0,0025 моля БФ в 10 мл свежего то-луола с добавкой в реакционную смесь 0,44г (0.1 моль/л) ДФХ. Полное исчерпывание исходного бисфенола БФ происходит через 5 минут.
Пример 11.
По примеру 1 окисляют 0,0025 моля БФ в 10 мл свежего то-луола, но в отсутствии катализатора КС. По истечении 60 минут от начала реакции получено 1г (100%) ДФХ.
Пример 12.
По примеру 1 окисляют 0,0025 моля БФ в 10 мл свежего толуола, но в отсутствии промотора Пж. По истечении 5 часов выделено 95% исходного БФ и 5% ДФХ.
Пример 13.
По примеру 1 окисляют 0,0025 моля БФ в 10 мл свежего то-луола в присутствии 1,3 г катализатора КС и 1% объем. (0,1 мл) Пж. Через 90 минут окисления выделили 1г (100%) ДФХ.
Пример 14.
Окисляют 0,0025 моля БФ в 10 мл свежего толуола в присутствии 1.3 г КС и 3.5% объем.(0.35мл) 20%-ного КОН. Выход ДФХ через 6 часов окисления кислородом воздуха при 85оС составил 20%.
Таблица 1.
(Т=85оС, Р=1атм., [БФ]=0,25 моль/л, Gкс=1,3г)
Пример 15 (прототип).
В автоклав объемом 300мл из нержавеющей стали загружается 2,21,6,61-тетратретбутил-4,41-бисфенол (5г), толуол (30мл), третбутиловый спирт (10мл) и 20%-ный раствор NаОН (5 капель). Автоклав закрывается, заполняется кислородом и нагревается до 75оС в течение часа при максимальном давлении 17.5 атм. Анализ реакционной смеси газожидкостным хроматографом показывает 83% конверсии 3,31,5,51-тетратрет-бутил-4.41-дифенохинона (ДФХ). Автоклав далее загружается 2,6-дитретбутил-фенолом (5г) и снова закрывается и нагревается до 75оС в течение 3 часов. После реакции автоклав открывается и реакционная смесь выгружается. Из охлажденной реакционной смеси выделяются кристаллы 2,21,6,61-тетратретбутил-4,41-бисфенола, часть которого возвращается на 1 стадию процесса /5/.
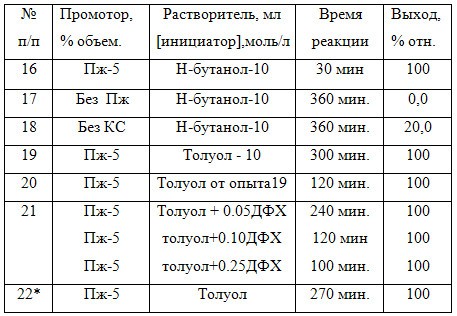
В примерах 16-22 и табл.2 приведены данные опытов по окислению 2.6-дитрет-бутил-фенола (2.6-ДТБФ) предлагаемым и известным способами.
Пример 16.
В емкость для окисления вводят 1,3 г катализатора КС*, 10 мл бутилового спирта, 1г (0.0046моль/л) 2,6-ДТБФ, и 5% объем. (0,5мл) Пж. Температуру реакционной смеси доводят до 85 оС и при этой температуре смесь перемешивают барботированием воздухом до полного исчерпывания 2,6-ДТБФ (30 минут). Отстоявшуюся горячую углеводородную фазу сливают с поверхности гетерогенного катализатора и Пж в стакан, где постепенно, по мере охлаждения, выпадает 1г темно-фиолетовых кристаллов ДФХ с Тпл.=246 оС, что соответствует литературным данным и составляет 100% от теоретического выхода.
Пример 17.
По примеру 16 окисляют 1г 2,6-ДТБФ в среде н-бутанола в отсутствие Пж. Окисление фенола в отсутствие Пж не идет. При нагревании в течение 6 часов выделен исходный фенол.
Пример 18.
По примеру 16 окисляют 1г 2,6-ДТБФ в среде н-бутанола в отсутствии фталоцианинового катализатора. При нагревании в течение 6 часов выделено 0,2 г ДФХ, что соответствует выходу 20%.
Пример 19.
По примеру 16 окисляют 1г 2,6-ДТБФ в среде толуола в течение 5 часов. Получают 1г (100%) ДФХ.
Пример 20.
По примеру 16 окисляют 1г 2,6-ДТБФ в среде возвратного толуола, отфильтрованного от кристаллов ДФХ от опыта 18. Полное исчерпывание исходного фенола и образование ДФХ со 100%-ным выходом наблюдается через 2 часа (вместо 5 часов по примеру 19).
Пример 21.
По примеру 16 окисляют 1г 2,6-ДТБФ в среде свежего толуола в присутствии добавок 0.05, 0.10 и 0.25 моль/л ДФХ. Время 100%-ной конверсии 2,6-ДТБФ составило соответственно 240, 120 и 100 мин.
Пример 22.
По примеру 16 окисляют 1г 2,6-ДТБФ в толуоле в присут-ствии 1,3г катализатора КСМ (взамен катализатора КС). Полное исчерпывание 2,6-ДТБФ с образованием ДФХ произошло через 4,5 часа.
Таблица 2
(КС*-1,3г, Р=1атм, [2,6-ДТБФ]=0,0046 моль/л, Т = 85оС)
*- во всех опытах использовался один и тот же образец катализатора КС.
**- опыт проводили в присутствии 1,3 г КСМ /8/.
Как видно из табл.1 и 2, проведение процесса окисления пространственно затрудненных фенолов предлагаемым способом позволяет значительно сократить продолжительность синтеза ДФХ и повысить его выход. Введение в реакционную смесь инициатора окисления — добавок синтезируемого ДФХ (примеры 8-10,21) или использование толуола-рециркулята с растворенным ДФХ (примеры 6,7,20), позволяет существенно ускорить процесс окисления как моно-, так и бисфенолов. В присутствии Пж окисление фенолов идет и без катализаторов КС и КСМ, но менее активно (примеры 11, 18).
Как видно из табл.1, при использовании рекомендуемого прототипом щелочного катализатора (вместо Пж) процесс окисления БФ идет со значительно меньшей скоростью и выходом ДФХ даже в комплексе с катализатором КС (пример 14) или, несмотря на высокое значение давления, использованное в примере 15 по прототипу. Предлагаемая каталитическая система является не только значительно более активной, по сравнению с прототипом, но и стабильной (табл.1, при-меры 1-5,14). Используемый в каталитической системе промотор окисления, обладая высоким удельным весом и деэмульгирующими свойствами, легко отделяется от углеводородного раствора продуктов реакции простым отстаиванием, практически не расходуясь в процессе синтеза. Легкость его отделения от углеводородного растворителя методом отстоя без применения дополнительных операций нейтрализации кислотами или водной отмывки позволяет резко сократить расходы реагентов и объемы стоков при получении замещенного дифенохинона и соответствующего бисфенола.
Непродолжительное время отстоя углеводородной и щелочной фаз позволяет существенно уменьшить размеры технологического оборудования, сократить величину капитальных и эксплуатационных затрат на реализацию процесса синтеза отечественных фенольных АОП.
Жесткое закрепление каталитически активного компонента на полимерном носителе катализатора исключает загрязнение синтезируемых продуктов нежелательными примесями каталитически активных металлов переменной валентности, и снижение активности катализатора в процессе эксплуатации, Это подтверждается данными анализа элементного состава и атомно-абсорбционных спектров ДФХ и БФ, синтезированных с помощью предложенной каталитической системы (в них отсутствуют примеси как фталоцианина кобальта, так и промотора), а также результатами многолетней промышленной эксплуатации катализатора КС в процессах окислительно каталитической демеркаптанизации углеводородного сырья /10/. В течение всего срока работы в водно-щелочной среде (8-10) лет катализатор КС сохранял свою первоначальную активность без подпитки и регенерации.
Источники информации
1. ЖПХ, т.58, №4, с.863.
2. Э.И. «Промышленный органический синтез», №3, 1976 г., реф.331.
3. Пат. США № 3.935.247, 1976, РЖ Хим. 1976, 17Н, 184П.
4. Пат. США № 3562338, 1971; №4238627, 1980, 568/730.
5. Пат США № 4.482.754, 1984г
6. А.С. № 2465621.
7. А.С. №1041142, 1983
8. Пат. РФ № 2110324, 1998
9. Пат. РФ № 2110555, 1998
10. Химия и технология топлив и масел, « 2, 1998, с.43
ФОРМУЛА ИЗОБРЕТЕНИЯ
1. Способ окисления пространственно затрудненных фенолов обработкой кислородом воздуха при нагревании в растворителях в присутствии окислительно-каталитической системы, ОТЛИЧАЮЩИЙСЯ тем, что в качестве окислительно-каталитической системы используют композицию, состоящую из фталоцианина кобальта и/или других водонерастворимых каталитически активных компонентов, жестко закрепленных на полимерном носителе катализатора, и/или из промотора окисления, являющегося продуктом превращения кислых примесей высококипящих углеводородных фракций при их окислительно-каталитической демеркаптанизации в щелочной среде, и из инициатора окисления, в качестве которого используют образующийся в этом процессе дифенохинон.
2. Способ по п.п.1, ОТЛИЧАЮЩИЙСЯ тем, что промотор используют в количестве не менее 1% от объема реакционного раствора.
3. Способ по п.1 и 2, ОТЛИЧАЮЩИЙСЯ тем, что дифенохинон вводят в реакционную систему в количестве от 0,1 до 100% (предпочтительно 3-30% от массы окисляемого фенола), либо непосредственно, либо в составе рециркулирующего растворителя.